We develop high-accuracy, low-scaling computational tools to inform, predict
and understand chemical and material systems at the most fundamental level.
Take a look around our site to learn more about what we do,
and feel free to get in contact with us to learn more.

Latest News
Website update and code release!
After four years of a stagnent website, I've finally updated the website! Having been through significant changes in group membership, many updates in our research directions/output, not to mention a global pandemic, I thought it was about time! In addition, we've just released our development code, called 'vayesta', which focuses on accurate and improvable approaches to ab initio quantum embedding. This has really been driven by Max, with important contributions from Charlie, Ollie, Alejandro and Basil. Hopefully it will be of wider use - check it out now by looking at our documentation here! Congratulations to all involved!
Opportunities
We are always on the lookout for motivated people to join the group! We currently have open positions for both PhD students and Postdocs.
See here for a currently open PDRA position.
See the How to Join page for more details about applying.
About
Who are we, and why is what we do interesting, possibly even important?! Well have a look at the About page for a lay-person's guide to what the fuss is about in computational electronic structure, and why we do what we do.
While you're at it, why not have a look at the Group page to see who 'we' all are!

Research
Our research focuses on a number of different areas of correlated electron phenomina, including molecular, solid-state and lattice problems. We see our techniques as resulting from a particularly fruitful blending of ab initio quantum chemistry and solid-state physics approaches, developing and applying techniques ranging from Monte Carlo, mean-field and machine learning, to Greens function methods. These are applied to understand and resolve unanswered questions about a wide variety of systems. We've also recently started increasing development of quantum computing algorithms for electronic structure too. See our Research page for more details on the developments in the group.
We sincerly thank funding bodies for their support of this research: The Royal Society, Air Force Office of Scientific Research and the European Research Council.
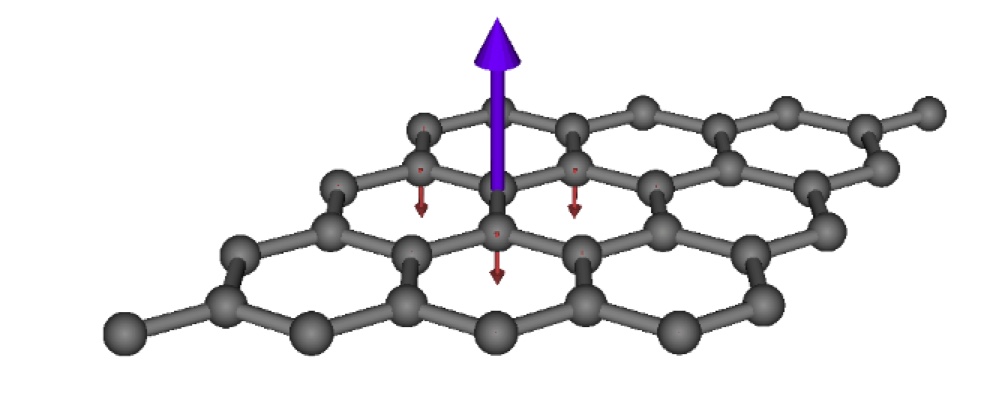
Research Highlight
Often, parameterizing a system via an efficient, compact, and polynomially-complex wavefunctions is something which can be done with relative ease. However, when it comes to optimizing these parameters using the variational principle, we have no option but to resort to stochastic, Monte-Carlo techniques. This can be exceptionally difficult when you are relying on noisy estimates at each point. With a PhD student, Lauretta Schwarz, we developed an improved approach, drawing on ideas from the supervised learning of neural networks, which is now published in Physical Review Letters entitled "Projector Quantum Monte Carlo Method for Nonlinear Wave Functions".
Above shows the short-ranged anti-ferromagnetic spin fluctuations in the ubiquitously prefixed 'wonder-material' Graphene, demonstrating the strength of the new approach

Where we are
Our research group is situated right in the heart of London, in King's College London. On the Strand, next door to the famous Somerset House, and overlooking the river Thames by Waterloo bridge.